Diseases
Click here to view more Diseases

Hereditary Tyrosinemia Type 1 (HT1) is a rare, autosomal recessive metabolic disorder characterized by a deficiency of the enzyme fumarylacetoacetate hydrolase (FAH). This enzyme plays a crucial role in the breakdown of tyrosine, an amino acid essential for normal growth and development. Without adequate FAH activity, toxic metabolites accumulate, leading to severe liver and kidney damage. HT1 is considered a medical emergency and requires early diagnosis and prompt intervention to prevent life-threatening complications.
HT1 has a global prevalence of approximately 1 in 100,000 to 1 in 120,000 live births. Although it is a rare disorder, there are geographic variations in its incidence. In India, the exact prevalence of HT1 remains unknown due to limited epidemiological data. However, with the increasing awareness and availability of diagnostic tools, the number of reported cases has been gradually rising.The genetic basis of HT1 in India is attributed to various mutations in the FAH gene. The most common mutation observed in Indian patients is c.1062+5G>A, followed by the missense mutation c.964G>A. However, the spectrum of mutations may vary across different regions of the country.
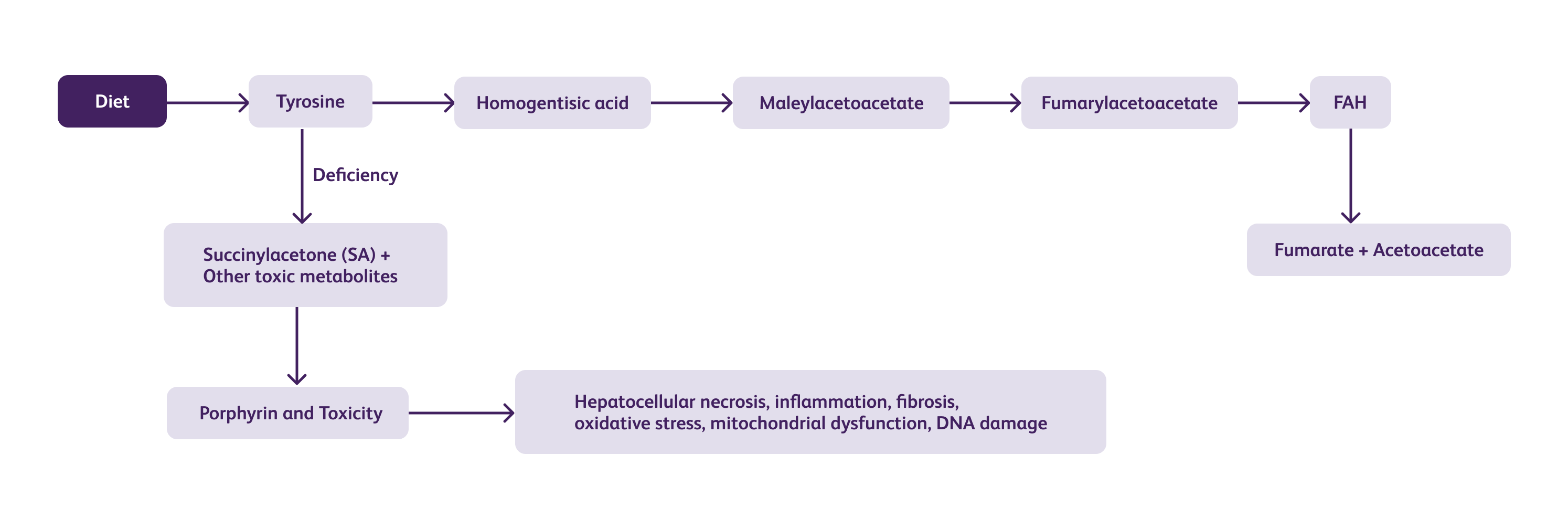
Tyrosine is an essential amino acid obtained from the diet.
In the liver, tyrosine is converted to 4-hydroxyphenylpyruvate (4-HPP) by the enzyme tyrosine aminotransferase (TAT).
4-HPP can enter two different pathways:
FAA generated in the minor pathway can also be metabolized to succinylacetone (SA) by the enzyme fumarylacetoacetate hydrolase (FAH).
In HT1, a mutation in the FAH gene leads to a deficiency or complete absence of FAH enzyme activity.
Without functional FAH, the major pathway of tyrosine degradation is disrupted.
The toxic metabolites can cross the blood-brain barrier and cause neurologic damage. Neurological symptoms may include intellectual disability, developmental delay, peripheral neuropathy, and seizures.
HT1 typically manifests within the first few months of life. HT1 exhibits a wide range of clinical manifestations, categorized into three forms based on the age of onset. The acute form typically emerges before six months of age, while the subacute form presents with initial symptoms between six and twelve months. The chronic form, on the other hand, occurs after twelve months of age. Among these forms, the acute variant is generally more severe, resulting in poor growth and often accompanied by liver damage. Unfortunately, infants with acute tyrosinosis may succumb to liver failure, leading to fatality within six to eight months if left untreated.
Hepatomegaly (enlarged liver) is commonly observed and may be present at birth or develop within the first few months of life.
Jaundice, characterized by yellowing of the skin and eyes, is a frequent finding.
Liver dysfunction can lead to coagulopathy (bleeding disorders) and a tendency to bruise easily.
Failure to thrive and poor weight gain are common in affected infants.
Ascites (accumulation of fluid in the abdominal cavity) may be present in severe cases.
Renal tubular dysfunction is a prominent feature in HT-1.
Renal Fanconi syndrome can develop, characterized by excessive excretion of amino acids, glucose, phosphate, and bicarbonate in the urine.
This can lead to polyuria (increased urine output), dehydration, and electrolyte imbalances.
Neurological crises can occur and are often triggered by a catabolic state, such as prolonged fasting or illness
Symptoms may include acute encephalopathy, seizures, hypotonia (decreased muscle tone), and developmental delay.
Peripheral neuropathy, characterized by numbness, tingling, and weakness in the limbs, can also occur.
Rickets: Due to impaired vitamin D metabolism and calcium absorption, rickets (softening and weakening of bones) may develop.
Hypophosphatemic rickets: In some cases, HT-1 can present with isolated hypophosphatemic rickets without obvious liver or renal involvement.
Hepatocellular carcinoma: Long-standing liver dysfunction and chronic inflammation increase the risk of hepatocellular carcinoma (liver cancer) in individuals with HT-1, particularly if not treated promptly.
The diagnosis of HT1 involves a combination of clinical suspicion, biochemical testing, and genetic analysis.
For infants developing symptoms before two months of age, the untreated one-year mortality rate is approximately 60%. To effectively screen for newborns, succinylacetone (SA) is the recommended primary marker due to its high sensitivity and specificity. Conversely, blood spot tyrosine levels lack both specificity and sensitivity. Although biochemical abnormalities can be detected shortly after birth, ideally within the first twelve hours, newborn screening allows for early intervention in asymptomatic infants. It is important to note that newborns diagnosed through screening may exhibit significantly elevated levels of AFP (α-fetoprotein) and mild coagulation irregularities.
When initially evaluating a suspected case of HT1, baseline tests should include the following assessments:
Blood gas analysis
Liver function tests: bilirubin, aspartate and alanine aminotransferase (AST, ALT), alkaline phosphatase, γ glutamyl transpeptidase (γGT), albumin
Coagulation profile: prothrombin time, partial thromboplastin time, fibrinogen
Urea and electrolyte levels, creatinine
Calcium and phosphate levels
Glucose and ammonia levels (in cases of acute liver failure)
Full blood count
Quantitative analysis of amino acids
α-fetoprotein (AFP) measurement
If available, succinylacetone (Note: plasma SA, being bound to proteins, is a more reliable test for monitoring metabolic control than urine SA, although urine SA is currently more widely accessible).
Glucose
Amino acids
Tubular re-absorption of phosphate (TRP)
Calcium/creatinine ratio
Albumin, protein, β2-microglobulin
Organic acids and succinylacetone (Note: Routine organic acid analysis may not be sufficiently sensitive)
Interpretation:
The initial tests may reveal indications of liver disease, often accompanied by significant clotting disorders. Raised levels of tyrosine and methionine in plasma amino acids may be observed, which are consistent with severe liver disease. Alpha-fetoprotein (AFP) levels are typically significantly elevated, although this marker is not specific to liver disease.
Ultrasound
Bone X-ray (recommended for individuals with confirmed tubulopathy): wrist orchest
MRI
Confirmatory diagnosis is established by detecting mutations in the FAH gene.
The management of HT1 requires a multidisciplinary approach involving hepatologists, metabolic specialists, dietitians, and liver transplant surgeons. The primary treatment modality is the restriction of tyrosine and phenylalanine in the diet, along with the administration of nitisinone (NTBC) (For more information Click Here), which inhibits the production of toxic metabolites. For patients with end-stage liver disease, liver transplantation is the only curative option.
Since tyrosine is an amino acid that cannot be effectively metabolized in HT-1, dietary restrictions on tyrosine-rich foods are necessary. Foods high in tyrosine include meat, fish, dairy products, eggs, legumes, nuts, and seeds. Phenylalanine, which is converted to tyrosine in the body, also needs to be restricted. Foods containing the artificial sweetener aspartame should be avoided as it is metabolized to phenylalanine.
Protein intake should be carefully controlled to ensure optimal growth and development while minimizing the accumulation of toxic metabolites. Protein sources low in tyrosine and phenylalanine, such as grains, vegetables, and fruits, should be emphasized. Specially formulated medical formulas may be prescribed to ensure adequate nutrition while limiting tyrosine and phenylalanine intake. These formulas are typically low in protein and contain essential nutrients, vitamins, and minerals.
Nitisinone is a potent inhibitor of the enzyme 4-hydroxyphenylpyruvate dioxygenase, which blocks the formation of toxic metabolites upstream of the FAH deficiency.
Nitisinone is the mainstay of pharmacological management in HT1 and has revolutionized the treatment of this disorder.
It effectively reduces the production of toxic metabolites, leading to improved clinical outcomes and a reduced risk of liver and renal complications.
Nitisinone therapy is initiated as early as possible, preferably within the first month of life. (For more information Click Here)
| Indication | Dose | Contraindications | Adverse Effects |
|---|---|---|---|
| Hereditary Tyrosinemia Type 1 (HT-1) |
The recommended starting dose is 0.5 mg/kg/day, given once daily. The maintenance dose is typically 0.1-1 mg/kg/day. Dosage may be adjusted based on individual patient response |
Hypersensitivity to nitisinone or any component of the formulation, severe liver impairment (Child-Pugh Class C). |
Common adverse effects include diarrhea, vomiting, abdominal pain, rash, and increased liver enzymes. Serious adverse effects include hepatocellular carcinoma (rare), liver failure (rare), and corneal opacities (rare). |
Liver transplantation remains an important therapeutic option for patients with advanced liver disease or hepatocellular carcinoma.
It can provide a definitive cure by replacing the deficient enzyme with a functional liver graft.
Liver transplantation may also be considered in patients who are non-compliant with dietary and medical management.
Challenges are non-adherence to dietary restrictions, limited availability of nitisinone (before nitisinone availability in India), and long-term complications. It explores potential future therapeutic strategies, including gene therapy, enzyme replacement therapy, and novel pharmacological interventions.
Hereditary Tyrosinemia Type 1 is a rare metabolic disorder with significant implications for affected individuals and their families. While the exact prevalence of HT1 in India remains uncertain, the number of reported cases is gradually increasing. Early diagnosis and appropriate management are essential for improving outcomes and preventing complications associated with liver and kidney dysfunction. Continued efforts in raising awareness, improving genetic testing capabilities, and expanding treatment options are crucial for addressing the challenges posed by HT1 in India.

Disclaimer
 Healthcare Professional
Healthcare Professional